MIT Think is calling for submissions to compete for its awards to find good research proposals that emphasize research and investigation into the background of a problem. I finished part of the writing on the motivations and Approach.
Motivation and Approach
By the end of 2021, COVID-19 has recorded more than 285 million confirmed cases and caused more than 5 million deaths globally. With waning immunity from vaccines and the emergence of new variants, there is still a huge need for effective drugs against the virus. (Giorgi et al., 2020; Singh et al., 2020) Drug companies have prioritized the development of such drugs, but their progress still is not fast enough. (The Drug Development Process, 2018) After a prospective drug is found, researchers must identify and minimize the risk of possible side effects, as well as maximize the drug’s disease-fighting potency. Candidate drugs must go through several rounds of animal and human trials, and even if they are proven to be safe and effective, patience is needed before mass production. (Pushpakom et al., 2019; The Drug Development Process, 2018) This entire process costs great amounts of time and resources. As shown in Figure 1, based on a report in 2018, introducing new drugs into the market requires 10 to 20 years and more than 2 billion US dollars. (Pushpakom et al., 2019; Wouters et al., 2020)
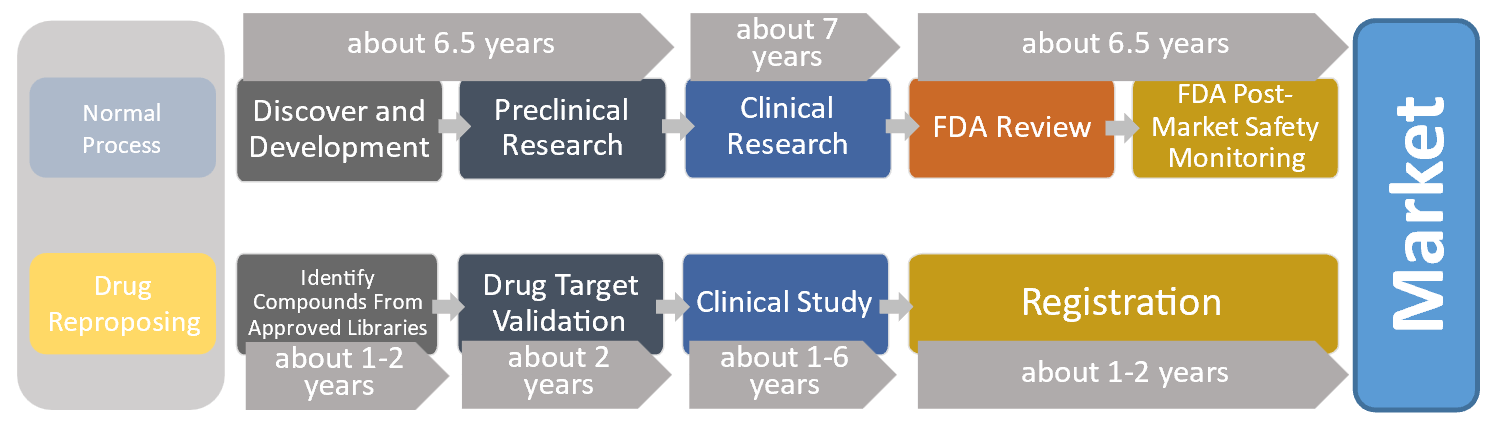
Figure 1. Drug Development Timeline of the normal process and the drug repurposing process, where the latter cost remarkably less development time.
Drug repurposing is possibly a game-changer. Following this strategy, researchers intend to find a cure by repurposing existing drugs that have been approved for other diseases. (Pushpakom et al., 2019) Since it involves the use of de-risked compounds, drug repurposing is expected to lower the overall development costs and shorten the development timelines. (Pushpakom et al., 2019; Sun et al., 2017) However, it remains a challenge to test a large pool of approved drugs for their effectiveness against the virus. In other words, drug repurposing serves to shorten the R&D cycle by reducing the risk factor, but identifying repurposable compounds from approved libraries, as shown in Figure 1, could still take 1 to 2 years. (Pushpakom et al., 2019; The Drug Development Process, 2018)
When researchers search for repurposable drug candidates, a key measure is binding affinity, which represents the strength of the binding interaction between a viral protein to an anti-virus drug. (Ballester & Mitchell, 2010; Singh et al., 2020) A stronger binding affinity means that the drug can inhibit the target protein more effectively than other compounds, suggesting a better chance of curing the infection. (Pushpakom et al., 2019) Thanks to breakthroughs in deep learning algorithms, drug-target interaction (DTI) prediction models are widely used to automate the searching process by scanning a large pool of existing drugs and predicting the interaction between drug molecular and protein sequence, promising to further shorten the development process. (Pushpakom et al., 2019)
The DTI models currently used for drug repurposing strategies can be classified into two categories. The first category includes Simboost (T. He et al., 2017), DTiGEMS+ (Thafar et al., 2020), DeepDTA (Öztürk et al., 2018), DeepCDA (Abbasi et al., 2020), and similar models based on random forest (RF) (Li et al., 2015) and support vector machine (SVM) (Li et al., 2015). These models rely on features extracted by human experts in some pre-processing stage and are typically fast in making DTI predictions, being able to process up to thousands of drug-target pairs per day (Li et al., 2015; Özçelik et al., 2021). However, since the extraction of features requires human expertise, the lack of it may lead to the loss of valuable information of the drug molecules and protein sequence, resulting in low prediction precision. (Özçelik et al., 2021)
The other type of models, such as Atomnet (Wallach et al., 2015) and SE-OnionNet(Wang et al., 2021), use 3-dimensional (3D) spatial structures of proteins and molecules for DTI predictions. While these models dramatically minimize the loss of information, they make predictions at a much slower rate, typically 1 drug-target pair per day. Moreover, in most databases, 3-D structure data are available for only a small fraction (0.2%-0.5%, depending on the database) of drug-protein pairs (Z. Liu et al., 2015). Due to data limitations, these models can only be applied to make DTI predictions in the specified database, whereas any real-world situation requires a model to be applied across different interaction mechanisms. (Wang et al., 2021) Low transferability is the main drawback of these models.
In this project, I intend to construct a model, which I call DeepPLA, that achieves high precision and high transferability, while at the same time producing predictions at a fast rate. Instead of 3-D structure data which are rarely available in databases, I propose to use 1-D protein sequence and molecular SMILES (Simplified Molecular Input Line Entry System) data. (Weininger, 1990) It may be a questionable approach to use 1-D protein sequence in medical research, but recent progress in computational models backs up this approach with strong outcomes. AlphaFold2 (The AlphaFold team, 2020), a model published by DeepMind, allows researchers to predict protein structure with sequence. The success of AlphaFold implies that we can make predictions based solely on the protein sequence. With this choice, my model can be trained with 20 to 500 times more data points for higher prediction accuracy, and the accessibility of new data points also makes my model applicable to targets previously ignored by state-of-the-art models, delivering higher transferability. (The AlphaFold team, 2020) To automate feature extraction, I will apply string-text-based embedding algorithms to ligand molecules and proteins separately, and feed the embedded numeric vectors into a stack of convolution neural networks and deep neural networks. Compared to the models like DeepDTA and DeepCDA (Abbasi et al., 2020; Öztürk et al., 2018), the string-text-based embedding algorithm conserves almost all of the information contained in the molecular and protein sequences, while lowering complexity. Finally, I will add Bi-LSTM layers that focus on partial interactions to capture detailed information.